
Case 56
Dr Marina-Portia Anthony / Professor Pek-Lan Khong
Clinical notes
A 44 year-old man with a history of thalassaemia presented for FDG-PET/CT to further investigate a lung mass incidentally found on a routine chest radiograph.Images
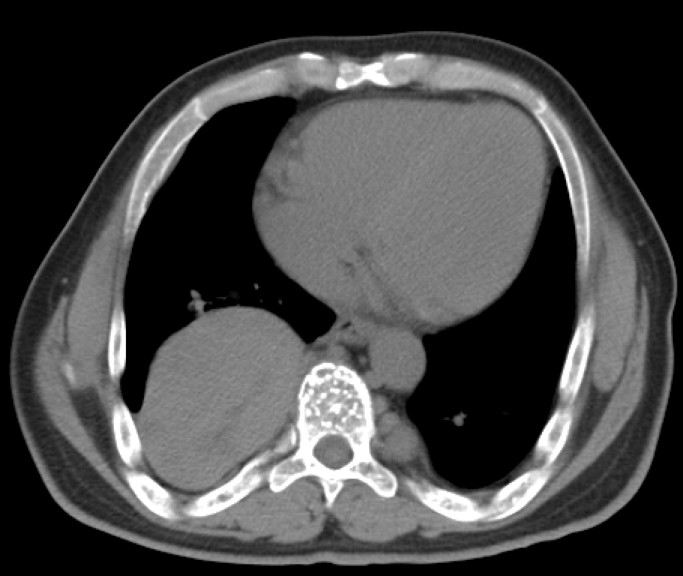

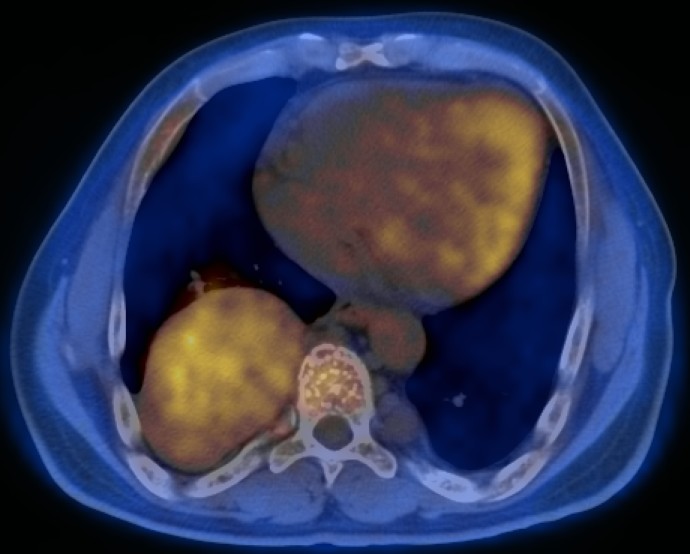
Figure 1.
Axial contrast-enhanced CT (A) and fused FDG-PET/CT (B) images through the mid-abdomen.
References
- Aflalo-Hazan V, Gutman F, Kerrou K, et al. Increased FDG uptake by bone marrow in major beta-thalassemia. Clin Nucl Med. 2005;30:754-5.
- Dunnick NR. Image interpretation session: 1999. Extramedullary hematopoiesis in a patient with beta thalassemia. Radiographics. 2000;20:266-8.
- Kumar V. Abbas A. Fausto N. Robbins and Cotran Pathologic Basis of Disease. 2005. Elselvier. Pennsylvania.
- Mosley C, Jacene HA, Holz A, Grand DJ, Wahl RL. Extramedullary hematopoiesis on F-18 FDG PET/CT in a patient with metastatic colon carcinoma. Clin Nucl Med. 2007;32(11):878-80.